JPCA:基于AIQM的高精度、低成本红外光谱模拟
近期,国际知名物理化学期刊《J Phys Chem A》发表了我们与厦门大学的合作研究工作。该工作探讨了如何基于AIQM实现高精度、高效率的红外光谱模拟计算,展示了人工智能增强量子化学计算的最新进展。该方法目前已被集成在MLatom的Aitomic插件中。

化学材料研究通常会涉及复杂的分子结构,红外光谱是解析化学结构的重要工具。通过量子化学方法如DFT等解析分子的红外光谱,往往耗时较长,计算成本高昂。而采用成本较低的半经验方法,则存在计算精度不足的问题。为了解决这一困境,我们引入了一系列全新的计算方法,即通用型机器学习AIQM方法。
通过与7000多个实验光谱数据比较,我们发现AIQM系列方法,尤其是最新开发的AIQM2方法,其红外光谱的模拟结果可以达到DFT方法的精度,而计算耗时仅与半经验方法GFN2-xTB相当,实现了更加高效快捷的气相红外光谱模拟。
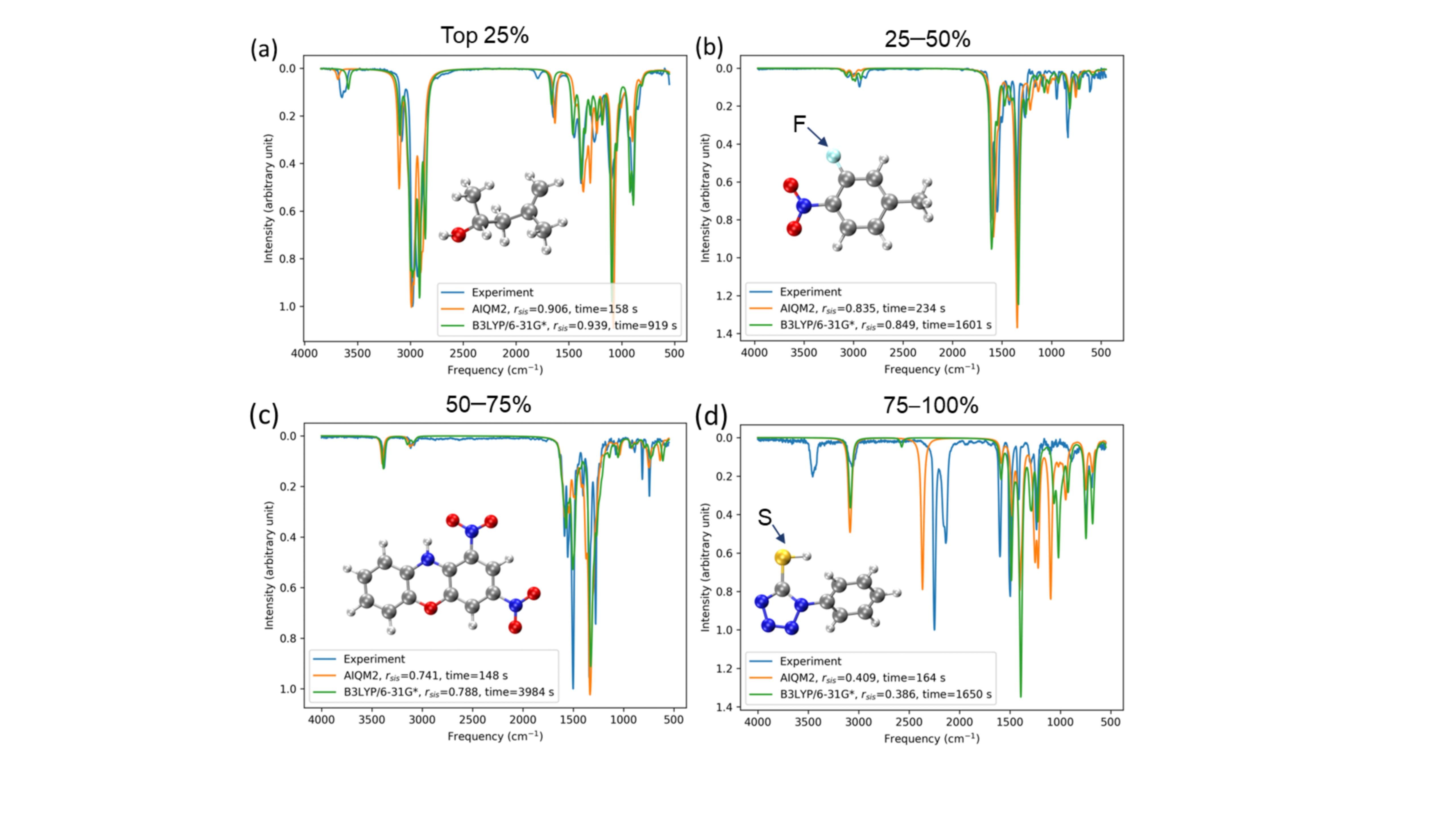
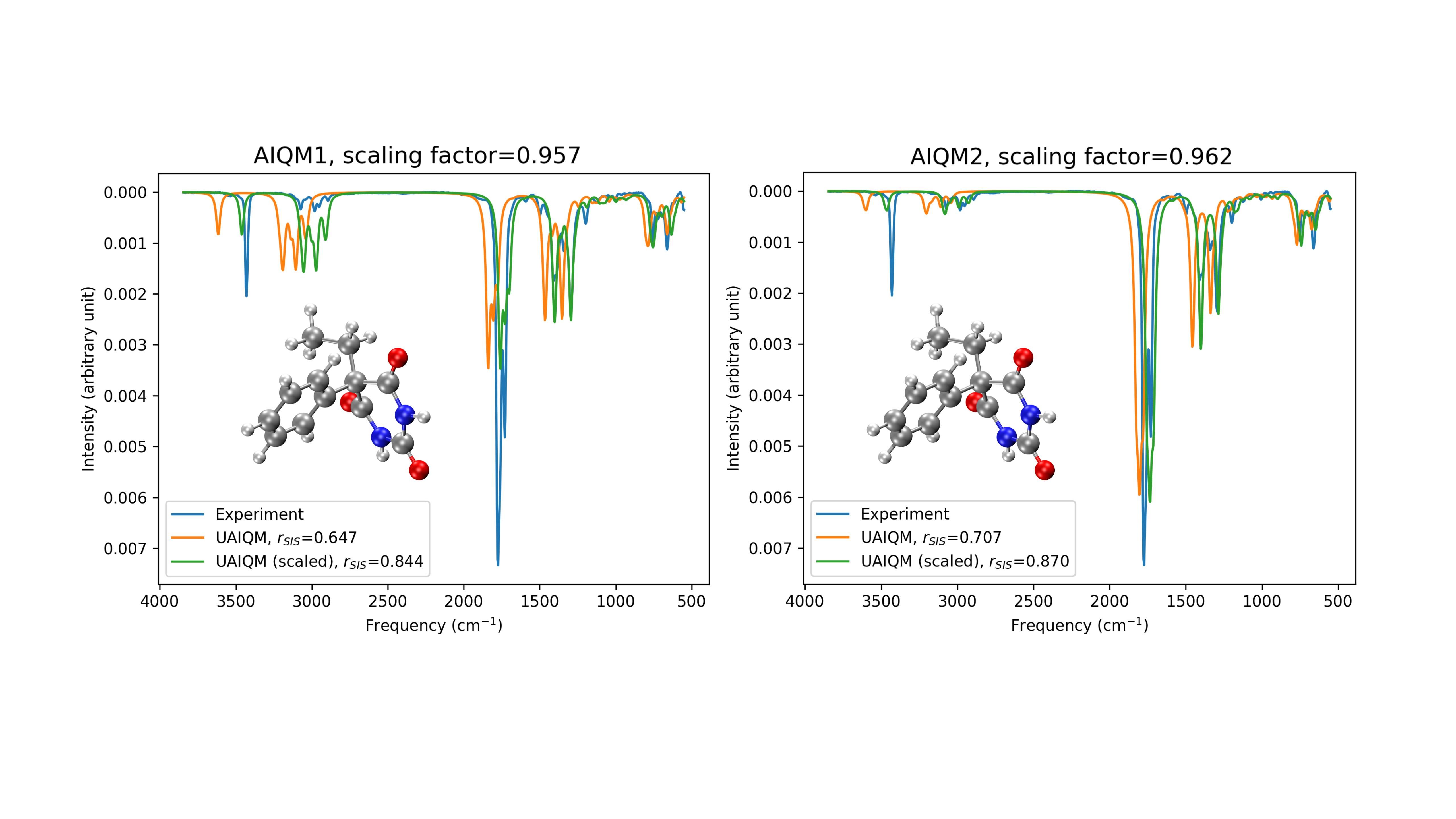
为了增强AIQM方法计算结果的可解释性,我们进一步引入经验参数,修正了分子振动频率,使模拟的红外光谱更好地贴合实验数据。
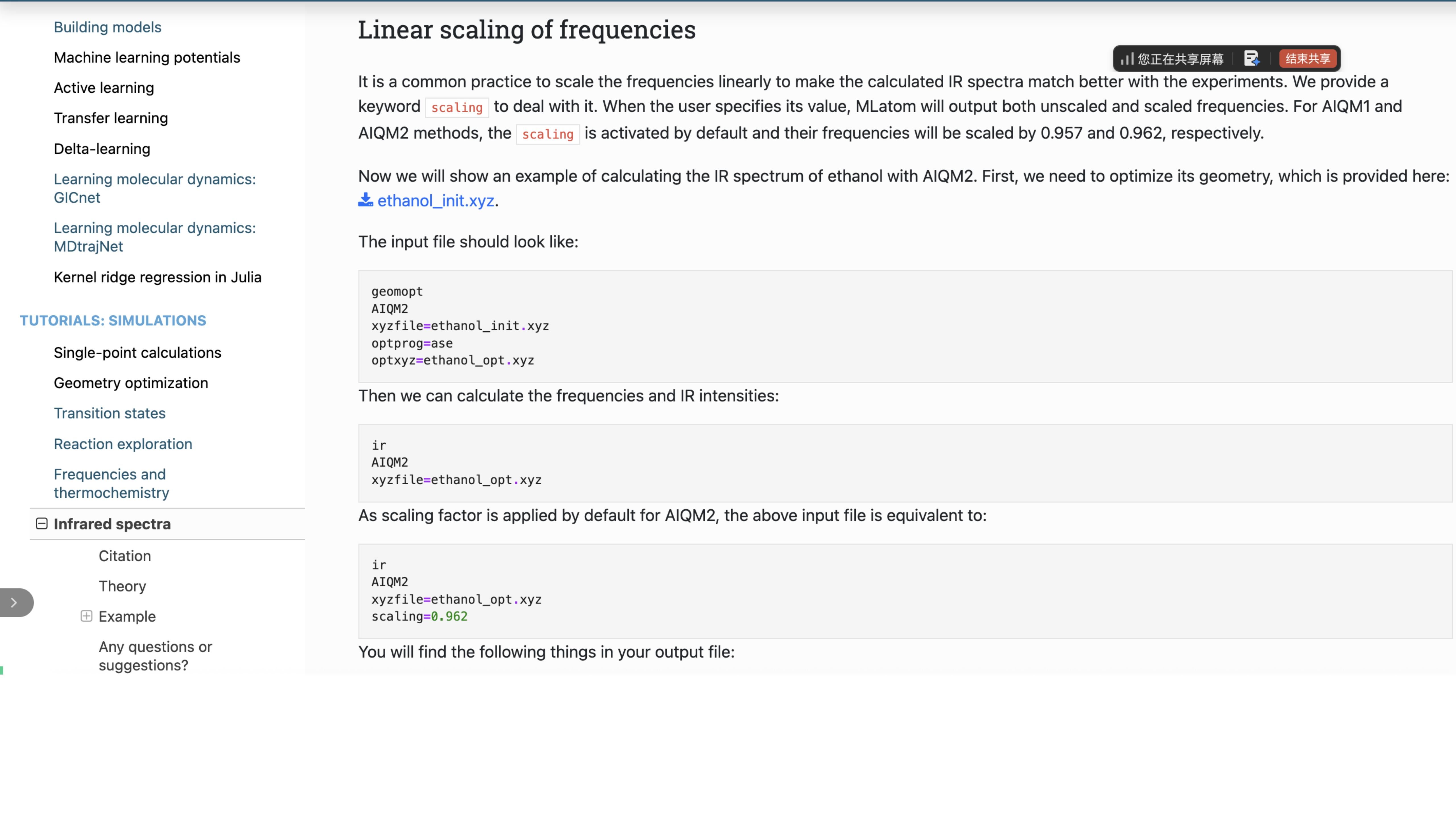
您可以参照MLatom的相关教程和演示脚本,轻松完成上述计算案例。
同时,您也可以登录Aitomistic Hub在线平台,体验高效便捷的红外光谱模拟计算。仅需三步(上传分子结构-确认分子结构-点击红外光谱按钮),系统就会为您自动执行AIQM2量子化学计算。Aitomistic Hub会实时显示计算结果,包括振动频率和强度等常规量子化学计算的完整数据。
值得一提的是,我们的计算流程对初始分子构型具有极强的容错能力,即使是明显不合理的分子构型(如下面视频演示的线性的己醇构型),AIQM2也能成功完成几何优化,得到正确的分子构型,然后再进行频率分析和红外光谱计算。
对于需要深入分析红外光谱计算结果的研究者,Aitomistic Hub还提供了Jupyter Lab集成环境。在这里,用户可以直观地观察红外光谱中每个特征峰对应的分子振动模式。
此外,您还可以在Aitomistic Hub上,通过与我们的人工智能助手Aitomia对话,完成红外光谱的模拟。如以下视频所展示,Aitomia在收到我们的需求后,自动完成了己醇分子建模、结构优化、频率计算和红外光谱模拟,计算结果与实验数据高度吻合。
如果您对上述计算操作有任何疑问,可以通过以下方式,查阅我们的教程指南、参考已发表的论文,或者加入QQ交流群在线咨询。