Aitomistic Updates: Conical Intersection Optimization, Langevin Thermostat MD, and New Active Learning Research
In this weekly briefing, you will learn about the aitomistic updates, including implementations in the newly released open-source MLatom 3.19.1, a new publication, and a preview of upcoming course updates and features for premium users.
Optimization of conical intersections
Starting from MLatom 3.19.1, which we released this week, you can perform optimization of the conical intersections. Mikolaj implemented the penalty-constraint approach of Levine et al. [J. Phys. Chem. B 112, 405–413 (2008)] and prepared a tutorial for you, which you can run on Aitomistic Hub as usual.
From this tutorial, you will learn that in this implementation, the geometry is optimized to minimize the average energy of two electronic states while penalizing the energy gap between them. It is an approximated method, which is particularly useful if you do not have nonadiabatic couplings (NACs). We have already used this method in our previous publication.
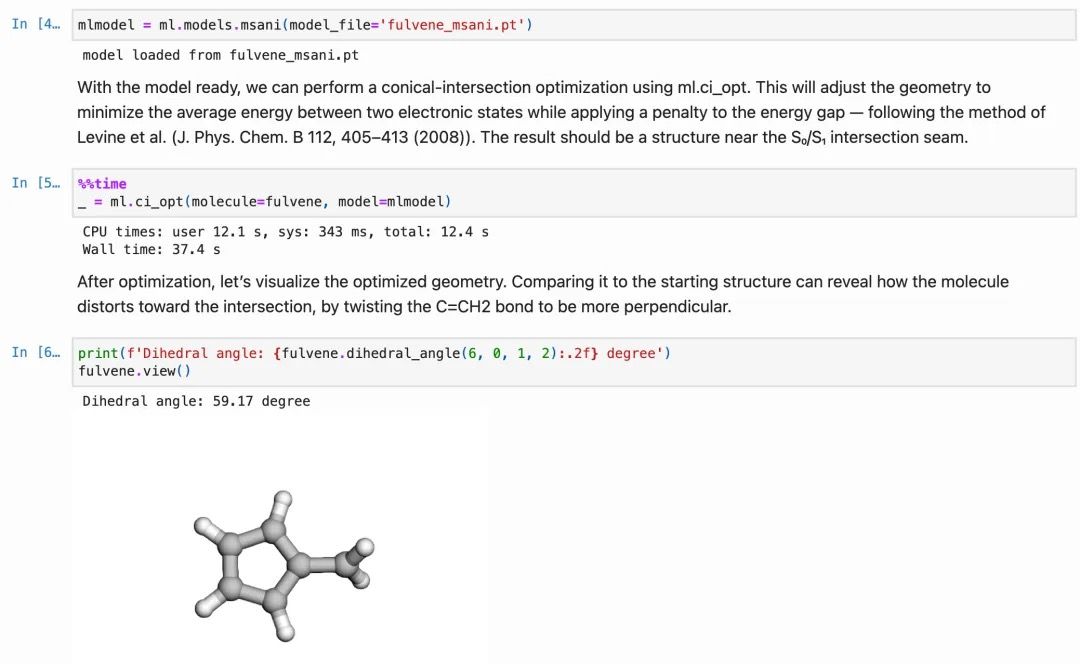
The tutorial shows two examples: optimization of conical intersection of fulvene with a pre-trained MS-ANI model and cyclohexadiene with a semi-empirical ODM2 method (the method I have developed when I was a post-doc in Max Planck Institute with late Walter Thiel). As usual with MLatom, you can visualize the optimized geometries and check their geometric parameters seamlessly in the Jupyter Notebook. Optimization of conical intersections is now as simple as:
ml.ci_opt(molecule=fulvene, model=mlmodel)Langevin thermostat
We keep improving open-source MLatom’s molecular dynamics engine, and Yi-Fan has included his implementation of the Langevin thermostat in the MLatom 3.19.1 release. You can do dynamics as usual by defining thermostat as, e.g.:
Langevin = ml.md.Langevin_thermostat(temperature=300,molecule=init_mol,tau=0.001)New publication on active learning with MLatom
Last Tuesday, a paper by Matheus de Oliveira Bispo and co-workers was published describing the MELTS program, interfacing Newton-X and MLatom to perform active learning to enable ML-accelerated surface-hopping dynamics. This program is of particular interest to the big Newton-X community.

Read more in the paper:
Matheus de Oliveira Bispo, Rafael Souza Mattos, Max Pinheiro Jr, Bidhan Chandra Garain, Pavlo O. Dral, Mario Barbatti. MELTS: Fully Automated Active Learning for Fewest-Switches Surface Hopping Dynamics. J. Chem. Theory Comput. 2025, ASAP.
Interested in the ins and outs of active learning?
Active learning for nonadiabatic dynamics on YouTube and Bilibili, based on paper with underlying methodology.
Active learning for ground-state simulations on YouTube and Bilibili, based on the physics-informed sampling protocol.
Upcoming updates for Premium users
Our Premium users enjoy early access to the updates. Among the new updates:
Fine-tuning of OMNI-P2x for your excited-state problem, enabling precise UV/vis spectra and more
AIQM4@DFT working for 14 elements
Fine-tuning of AIQM models for your tasks
Updated living course on computational chemistry and AI, with webinars
More tokens for autonomous simulations with our Agents
More computational time
Interested in becoming a Premium user? Please contact us!
Please also let us know what you would like us to improve to make your experiences with Aitomistic better. Simply reply to this email or join us on Slack to directly chat with us.